

Ph.D. - 1998 - University of Alberta
Postdoc - 1999-2002 - Harvard Medical School with Peter T. Lansbury, Jr.
Currently Accepting Students
A book that Dr. Rochet has co-edited, entitled ‘Oxidative Stress and Redox Signalling in Parkinson’s Disease’, has just been published by the Royal Society of Chemistry (DOI 10.1039/9781782622888). Dr. Rochet and his graduate students wrote one of the chapters in this book (Chapter 11). For additional details, please see:
https://pubs.rsc.org/en/content/ebook/978-1-78262-188-1
Research in the Rochet laboratory is aimed at understanding the role of protein aggregation in neurodegenerative disorders, with an emphasis on Parkinson's disease (PD). PD, an age-related neurodegenerative disorder that disrupts the lives of an estimated 5 million people worldwide, is manifested by classic motor symptoms including resting tremor, slowness of movement, rigidity, and postural instability. These symptoms result largely from a loss of dopaminergic neurons from the substantia nigra in the midbrain, and this neuronal loss is thought to involve oxidative stress and aggregation of the presynaptic protein α-synuclein (aSyn). Current therapies only temporarily relieve symptoms without slowing the underlying neurodegenerative disease. In addition, a large proportion of neurons have been destroyed by the time PD symptoms are detectable, and no therapies exist to reverse this damage. Accordingly, there is a critical need for neuroprotective strategies to help reduce the risk of PD.
Two key observations suggest that aSyn aggregation plays a role in PD pathogenesis. First, a characteristic feature of PD brain is the presence in some surviving neurons of Lewy bodies, cytosolic inclusions enriched with fibrillar forms of aSyn. Second, autosomal-dominant mutations in the aSyn gene have been identified in patients with early-onset familial PD, and evidence suggests that these mutations promote aSyn aggregation. Despite this evidence, the molecular details underlying aSyn neurotoxicity in PD are unclear.
Data obtained by our group and others suggest that aSyn aggregation is promoted by post-translational modifications resulting from mitochondrial dysfunction/oxidative stress and by binding of the protein to phospholipid membranes. aSyn self-assembly involves the initial formation of early aggregates (oligomers and ‘protofibrils’) which then convert to amyloid-like fibrils of the type found in Lewy bodies. Various lines of evidence suggest that aSyn oligomers or protofibrils may be more neurotoxic than mature fibrils, although this remains an open question in the PD field. A number of neuroprotective proteins prevent the accumulation of toxic, aggregated forms of aSyn in the brain, including: (i) proteins with antioxidant functions, (ii) molecular chaperones, and (iii) proteins that play a role in cellular clearance mechanisms such as the ubiquitin-proteasome system and lysosomal autophagy. In addition to our studies of aSyn, a number of projects in the lab are focused on DJ-1, a neuroprotective protein that is dysfunctional in familial and sporadic forms of PD and is involved in each of the cellular defense mechanisms outlined above.
Model illustrating cellular phenomena that promote or inhibit a-Syn self-assembly. A loss of mitochondrial function (e.g., impairment of complex I) or an increase in cytosolic dopamine levels triggers a buildup of ROS in dopaminergic neurons. In turn, ROS and/or dopamine oxidation products react with a-Syn, converting the protein to oxidized forms with a high propensity to aggregate. a-Syn self-assembly is promoted under some conditions by binding of the protein to phospholipid membranes. a-Syn aggregation and neurotoxicity may be mitigated by (i) cellular clearance mechanisms, including the 26S proteasome and lysosomal autophagy; (ii) cellular antioxidant responses, including Nrf2-mediated transcription, resulting in increased glutathione (GSH) synthesis, and MsrA-dependent repair of oxidized a-Syn; and (iii) molecular chaperones, including Hsp70, Hsp27, aB-crystallin, and DJ-1. Adapted from Rochet, J.-C.*, Hay, B. A., and Guo, M.* (2012) Molecular insights into Parkinson's disease, Prog. Mol. Biol. Transl. Sci. 107, 125-188.
Our research is aimed at better understanding mechanisms of neurotoxicity and neuroprotection in PD, with the ultimate goal of identifying new therapeutic strategies. Examples of questions addressed by our studies include:
- What are molecular mechanisms (e.g. intermolecular interactions, conformational changes) underlying the formation of neurotoxic aSyn aggregates?
- How do post-translational modifications (e.g. oxidation, phosphorylation, nitration) modulate aSyn aggregation and neurotoxicity?
- What are cellular factors (e.g. antioxidant proteins, chaperones, proteins involved in lysosomal autophagy) that modulate aSyn aggregation and neurotoxicity?
- Which cellular functions (e.g. mitochondrial respiration, lysosomal autophagy) are disrupted by neurotoxic forms of aSyn?
- Can we identify chemical entities that interfere with aSyn aggregation or neurotoxicity or that enhance the neuroprotective function of DJ-1?
These questions are addressed using an interdisciplinary approach spanning various methodologies, including:
- Protein biochemistry and cell biology (e.g. circular dichroism, fluorescence, ultracentrifugation, light scattering, confocal microscopy, flow cytometry)
- Site-directed and random mutagenesis
- Characterization of neurotoxic or neuroprotective mechanisms in mammalian cells, primary midbrain cultures, and in vivo rodent models
- Behavioral and immunohistochemical analysis of rats injected in the brain with viral vectors encoding proteins with neurotoxic effects (e.g. alpha-synuclein) or neuroprotective activity (e.g. endosulfine-alpha)
- Biochemical and immunohistochemical analysis of post-mortem human brain tissues
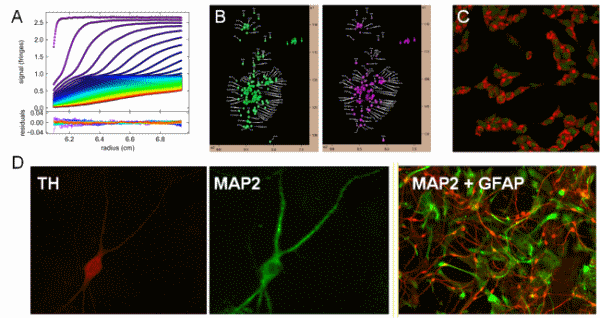
Images representing different experimental approaches used in the Rochet lab. (A) Sedimentation-velocity data obtained by ultracentrifugation of recombinant proteins. (B) HSQC-NMR data obtained for wild-type aSyn in the absence (left) and presence (right) of phospholipid vesicles. (C) Image of SH-SY5Y neuroblastoma cells stained with the lysosome-specific dye acridine orange. (D) Images of a rat primary midbrain neuron stained for the dopaminergic marker tyrosine hydroxylase (TH) (left); the same neuron stained for the general neuronal marker MAP2 (center); and a primary midbrain culture stained for MAP2 (red) and the glial marker GFAP (green) (right).
Our research is enhanced through collaborations with groups in MCMP and other departments at Purdue University, in addition to groups at other institutions throughout the world. Examples of collaborations with colleagues in MCMP and at Purdue are shown below.
Gideon Drafor (Graduate Student)
Serina Ayse Gogusoglu (Graduate Student)
Wenzhu Qi (PULSE Graduate Student)
Dem X Santiago Santiago (Graduate Research Assistant)
Alicia Nicole Scott (Graduate Student)
Protein misassembly in neurodegenerative disorders
e.g. α-synuclein aggregation in PD, dementia with Lewy bodies
Protein-lipid interactions in amyloid diseases
e.g. effects of phospholipid membranes on α-synuclein aggregation, toxicity
Post-translational modifications in aging and age-related neurodegenerative disorders
e.g. oxidation, phosphorylation of α-synuclein; oxidation of DJ-1
Mitochondrial proteins in age-related neurodegenerative disorders
e.g. DJ-1, Omi/HTRA2, PGC1α
Antioxidant response pathways in aging and age-related neurodegenerative disorders
e.g. pathways involving DJ-1, MsrA, Nrf2, glutathione
Molecular chaperones in aging and age-related neurodegenerative diseases
e.g. DJ-1, Hsp27, Hsp70
Autophagy in aging and age-related neurodegenerative diseases
Intracellular transport mechanisms in neurodegenerative disorders
e.g. ER-Golgi trafficking, aggresome formation
Small-molecule inhibitors of protein aggregation, neurodegeneration
e.g. sirtuin 2 inhibitors, retinoids/rexinoids
Dietary components as inhibitors of protein aggregation, neurodegeneration
e.g. riboflavin, blueberry extract, soy isoflavones
Glia-neuron interactions in neurodegenerative disorders
Mechanisms of neurodegeneration associated with methamphetamine intoxication
Epigenetic changes in CNS disorders
de RUS Jacquet*, A., Tambe, M. A., Ma, S. Y., McCabe, G. P., Vest, J. H. C., and Rochet, J.-C.* (2017) Pikuni-Blackfeet traditional medicine: neuroprotective activities of medicinal plants used to treat Parkinson’s disease-related symptoms. J. Ethnopharmacol., in press. *Co-corresponding authors.
Ysselstein, D., Dehay, B., Costantino, I. M., McCabe, G. P., Frosch, M. P., George, J. M., Bezard, E., and Rochet, J.-C. (2017) Endosulfine alpha inhibits membrane-induced α-synuclein aggregation and protects against α-synuclein neurotoxicity. Acta Neuropathol. Commun. 5, 3. Full Text (HTML)
Ysselstein, D., Joshi, M., Mishra, V., Griggs, A. M., Asiago, J. M., McCabe, G. P., Stanciu, L. A., Post, C. B., and Rochet, J.-C. (2015) Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol. Dis. 79, 150-163. Full Text (HTML)
Griggs, A. M., Agim, Z. S., Mishra, V. R., Tambe, M. A., Director-Myska, A. E., Turteltaub, K. W., McCabe, G. P., Rochet, J.-C., and Cannon, J. R. (2014) 2-Amino-1-Methyl-6-Phenylimidazo[4,5-b]Pyridine (PhIP) is Selectively Toxic to Primary Dopaminergic Neurons In Vitro. Toxicol. Sci. 140, 179-189. Abstract | Full Text (HTML) | Full Text (PDF)
Strathearn, K. E., Yousef, G. G., Grace, M. H., Roy, S. L., Tambe, M. A., Ferruzzi, M. G., Wu, Q.-L., Simon, J. E., Lila, M. A., and Rochet, J.-C. (2014) Neuroprotective effects of anthocyanin- and proanthocyanidin-rich extracts in cellular models of Parkinson’s disease. Brain Res. 1555, 60-77. Abstract | Full Text (HTML) | Full Text (PDF)
Tardiff, D. F., Jui, N. T., Khurana, V., Tambe, M. A., Thompson, M. L., Chung, C. Y., Kamadurai, H., Kim, H.-T., Lancaster, A. K, Caldwell, K. A., Caldwell, G. A., Rochet, J.-C., Buchwald, S. L., and Lindquist, S. (2013) Phenotypic screening and chemical genetics reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity. Science 342, 979-983. Abstract | Full Text (HTML) | Full Text (PDF)
Rochet, J. C.*, Hay, B. A., and Guo, M.* (2012) Molecular insights into Parkinson's disease, Prog. Mol. Biol. Transl. Sci.107, 125-188. *Co-corresponding authors. Abstract | Full Text (HTML) | Full Text (PDF)
Madian, A. G., Hindupur, J., Hulleman, J. D., Diaz-Maldonado, N., Mishra, V. R., Guigard, E., Kay, C. M., Rochet, J.-C.*, and Regnier, F. E.* (2012) Effect of single amino acid substitution on oxidative modifications of the Parkinson’s disease-related protein, DJ-1. Mol. Cell. Proteomics 11(2):M111.010892. *Co-corresponding authors. Abstract | Full Text (HTML) | Full Text (PDF)
Dumitriu, A., Pacheco, C. D., Wilk, J. B., Strathearn, K. E., Latourelle, J. C., Goldwurm, S., Pezzoli, G., Rochet, J.-C., Lindquist, S., & Myers, R. H. (2011) Cyclin-G associated kinase modifies α-synuclein expression and toxicity in Parkinson’s disease: results from the GenePD Study. Hum. Molec. Genet. 20:1478-1487. Abstract | Full Text (HTML) | Full Text (PDF)
Zheng, B., Liao, Z., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., Eklund, A. C., Zhang-James, Y., Kim, P. D., Hauser, M. A., Grünblatt, E., Moran, L. B., Mandel, S. A., Riederer, P., Miller, R. M., Federoff, H. J., Wüllner, U., Papapetropoulos, S., Youdim, M. B., Cantuti-Castelvetri, I., Young, A. B., Vance, J. M., Davis, R. L., Hedreen, J. C., Adler, C. H., Beach, T. G., Graeber, M. B., Middleton, F. A., Rochet, J.-C., & Scherzer, C. R., the Global PD Gene Expression (GPEX) Consortium. (2010) PGC-1α is a therapeutic target for early intervention in Parkinson’s disease Sci. Transl. Med. 2(52):52ra73. Abstract | Full Text (HTML)
Haque, F., Pandey, A. P., Cambrea, L. R., Rochet, J.-C.*, & Hovis, J. S.* (2010) Adsorption of α-synuclein on lipid bilayers: modulating the structure and stability of protein assemblies. J. Phys. Chem. B. 114:4070–4081. *Co-corresponding authors. Abstract | Full Text (HTML) | Full Text (PDF)
Gitler, A. D., Chesi, A., Geddie, M. L., Strathearn, K. E., Hamamichi, S., Hill, K. J., Caldwell, K. A., Caldwell, G. A., Cooper, A. A., Rochet, J.-C., & Lindquist, S. 2009. Alpha-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 41:308-315. Abstract | Full Text (HTML) | Full Text (PDF)
Pandey, A. P, Haque, F., Rochet, J.-C.*, & Hovis, J. S.* 2009. Clustering of alpha-synuclein on supported lipid bilayers: role of anionic lipid, protein, and divalent ion concentration. Biophys. J. 96:540-551. *Co-corresponding authors. Abstract | Full Text (HTML) | Full Text (PDF)
Liu, F., Hindupur, J., Nguyen, J. L., Ruf, K. J., Zhu, J., Schieler, J. L., Bonham, C. C., Wood, K. V., Davisson, V. J., & Rochet, J.-C.. (2008) Methionine sulfoxide reductase A protects dopaminergic cells from Parkinson's-related insults. Free Radic. Biol. Med. 45: 242-255. Full Text (HTML) | Full Text (PDF)
Liu, F., Nguyen, J. L., Hulleman, J. D., Li, L., & Rochet, J.-C.. (2008) Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson's disease. J. Neurochem. 105: 2435-2453. Abstract | Full Text (HTML) | Full Text (PDF)